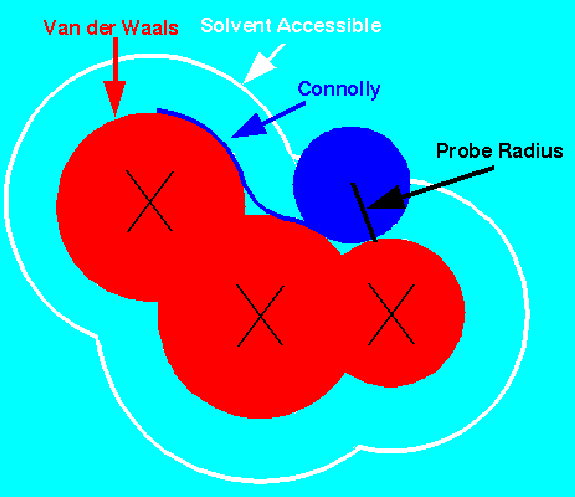
A common representation is due to Connolly [2]. It virtually rolls a
'water' probe ball (1.4-1.8 Å diameter) over the Van der Waals surface,
smoothing the surface and bridging narrow crevices, which are inaccessible to the solvent.
This partitions the surface into convex, concave and saddle patches according
to the number of contact points between the surface atoms and the probe ball.
As Output, the representation consists of points + normals to the surface.
These are sampled according some required sampling density (e.g. 10 pts/Å2).
 |
 |
There are different ways to represent shape complementarity. The points + normals representation [2] is a dense one. The points representation in [10] is sparser, and so is the Solid Angle local extrema [1]. Another representation is SPHGEN [5] - surface cavity modeling by pseudo-atom centers.
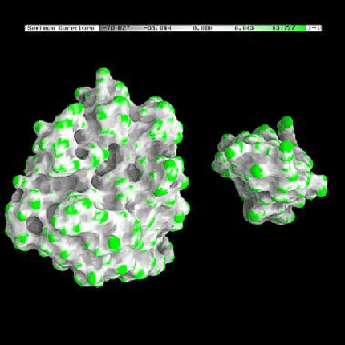
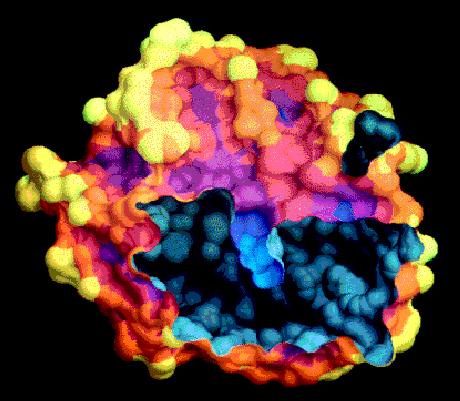
One of the advantages of the molecular surface is its ability to visualize the shape complementarity at interfaces, as show on figures 13.12 and 13.13.
 |
 |
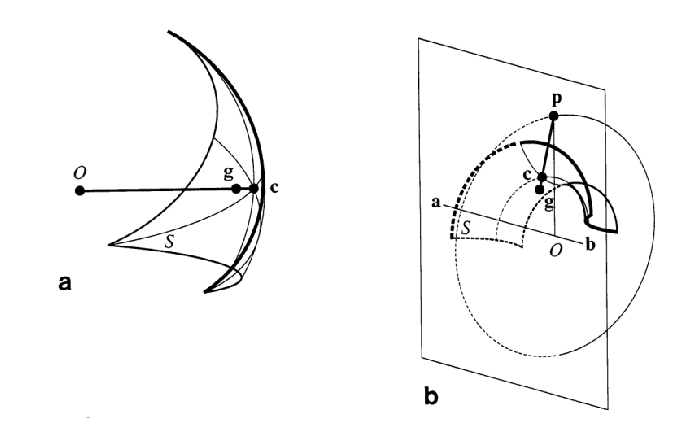
In the critical points representation based on Connolly's representation we define a single point and a normal for each patch, as illustrated in figure 13.14. Convex patches are called caps, concave ones - pits and saddles are called belts.
 |
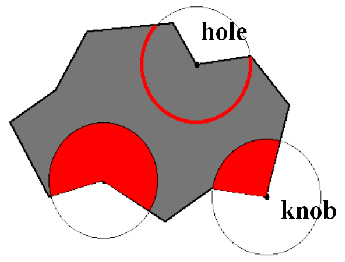
The Solid Angle local extrema (knobs - holes) representation is based upon centering a sphere at the protein surface and measuring the fraction of the sphere inside the solvent-excluded volume of the protein. If more than half of the sphere is inside the protein, the region is concave, if less than half of the sphere is inside the protein, the shape is convex. Either the solid sphere or the sphere surface may be used. A two-dimensional example is shown in figure 13.15.
 |
|
The SPHGEN representation generates sets of overlapping spheres to describe the shape of a molecule or molecular surface. For receptors, a negative image of the surface invaginations is created; for a ligand, the program creates a positive image of the entire molecule. Each sphere touches the molecular surface at two points and has its radius along the surface normal of one of the points. For the receptor, each sphere center is "outside" the surface, and lies in the direction of a surface normal vector. For a ligand, each sphere center is "inside" the surface, and lies in the direction of a reversed surface normal vector. Spheres are calculated over the entire surface, producing approximately one sphere per surface point. This very dense representation is then filtered to keep only the largest sphere associated with each receptor surface atom. The filtered set is then clustered on the basis of radial overlap between the spheres using a single linkage algorithm. This creates a negative image of the receptor surface, where each invagination is characterized by a set of overlapping spheres. These sets, or "clusters", are sorted according to numbers of constituent spheres, and written out in order of descending size. The largest cluster is typically the ligand binding site of the receptor molecule.