


Next: Results on Real DNA
Up: Constructing Physical Maps from
Previous: Map Quality
Results presented in this section are for the base scenario, which
has the following parameters:
- Length of clones:
lc = 40960 base pairs.
- Target genome length: L = 25 clone lengths (
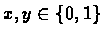 which is approximately
1M base pairs).
which is approximately
1M base pairs).
- Clone coverage: 10
- False negatives probability:
- False positives probability:
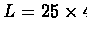
- Number of probes: n = 500
- Length of probes: plen = 8
Based on the results of 1000 simulations, the algorithm has a
probability of
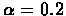 of making any big errors in the
base scenario. The average error in the constructed map is also
quite small:
of making any big errors in the
base scenario. The average error in the constructed map is also
quite small:
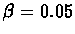 The average error can be reduced if
a finer quantization unit is used (at a linear cost to memory and
CPU consumption). A further experiment indicated a probability of
about 0.075 of making any mistake that exceeds a clone's length in the
estimation of the relative distance between any two (not
necessarily adjacent) clones.
The average error can be reduced if
a finer quantization unit is used (at a linear cost to memory and
CPU consumption). A further experiment indicated a probability of
about 0.075 of making any mistake that exceeds a clone's length in the
estimation of the relative distance between any two (not
necessarily adjacent) clones.
Figure:
True clone order (y
axis) vs. constructed order (x axis) in four scenarios. When
applicable, weak points shown as vertical dotted lines. The
results are taken from the following scenarios: (a) base scenario,
(b) a long 2MB genome, (c) a simulation with very low coverage
(5), (d) a simulation with very low coverage (5) and very high
noise (
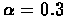 and
and
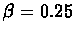 ). Note that all big
errors were detected as weak points, though some weak points
incorrectly suggested additional big errors. pinpoint possible
errors. This information can be used for a judicious choice of
additional hybridization experiments, minimizing cost and human
effort.
). Note that all big
errors were detected as weak points, though some weak points
incorrectly suggested additional big errors. pinpoint possible
errors. This information can be used for a judicious choice of
additional hybridization experiments, minimizing cost and human
effort.
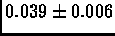 |
Figure 9.13:
Influence of various
simulation parameters on the probability of having big errors. The
vertical dotted line indicates the value of the parameter in the
base scenario. Note that the effect of a decrease in the number of
probes is very similar to that of an increase in the experimental
noise. This is because noise decreases the informational content
of each probe, an effect that can be countered by an increase in
the number of probes. It is also notable that the probe size has a
very significant effect, resulting from its direct influence on
the frequency of probe occurrences, and therefore on the
informational content of the experiment. In contrast, the genome
size only moderately effects performance.
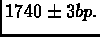 |
In analogy to the breaking up of a chemical molecule, the
separation of a contig into two nonoverlapping parts should
increase the energy substantially. However, if the two parts do
not overlap in the real map, the separation energy should be quite
small or even negative.

Such information can be used by the laboratory in order to
pinpoint areas where additional hybridizations should be
performed. We also make use of the weak points in our algorithm in
order to break up a contig and reassemble it. In case an error was
made at an early stage, this process enables the algorithm to
correct its previous error with the benefit of the additional
information from other clones added at a later stage. An example of weakpoint detection is described in Figure 9.12.
Next: Results on Real DNA
Up: Constructing Physical Maps from
Previous: Map Quality
Peer Itsik
2001-01-09