


Next: FastA - Steps
Up: Tools for Searching
Previous: Tools for Searching
FastA is a sequence comparison software that uses the method of Pearson and Lipman [5].
The basic FastA algorithm assumes a query sequence and a database over the same alphabet. It searches a DNA sequence in a DNA database or a protein sequence in a protein database. Practically, FastA is a family of programs, allowing also queries of DNA vs. a protein database, or vice versa.
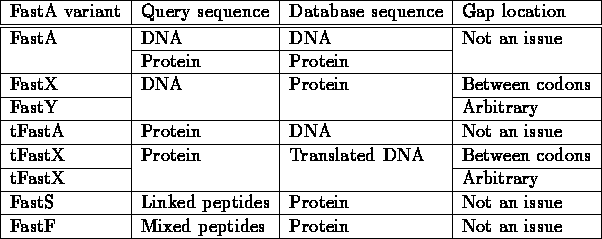
In these variants there is further distinction, which regards the location of gaps: one may assume that gaps occur only in the codon frames corresponding to amino-acid insertion; alternatively, one can assume gap location to be arbitrary, accounting for insertion/deletion of nucleotides.
The different FastA variants are summarized in the following table:
Figure 5.2:
Vairants of FastA algorithm
 |
Under different circumstances it is favorable to use different programs:
- To identify an unknown protein sequence use either: FastA3, Ssearch3 or tFastX3
- To identify structural DNA sequence: (repeated DNA, structural RNA) use FastA3, first with ktup=6 and than with ktup=3.
- To identify an EST use FastX3 (check whether the EST codes for a protein homologous to a known protein).
- Use ktup=1 for oligonucleotides (length < 20).
- Search speed and selectivity are controlled with the ktup (word-size) parameter. Searches with ktup=1 are slower, but more sensitive, while ktup=2 is faster but less effective.
- For proteins, the default is ktup=2. For DNA, the default is ktup=6. Use ktup=3 or ktup=4 more sensitivity.
FastA3 (Fastx3, etc.) is the current version of FastA. Tha main important over version 2 (which is still in common use) is a much better normalization method for alignment scores.
FastA is available directly via the FastA3 server, [11] or it can be accessed through one of the retrieval systems e.g. the GenWeb mirror site at the Weizmann Lustitude.[7]
Next: FastA - Steps
Up: Tools for Searching
Previous: Tools for Searching
Peer Itsik
2000-12-11