


Next: Programs in the FastA3
Up: The FastA Software Package
Previous: Variants of FastA
Sketch of the FastA Algorithm (see also lecture No. 3)
FastA locates regions of the query sequence and matching regions in the database sequences that have high densities of exact word matches. The score for such a pair of regions is saved as the init1 score. Then FastA determines if any of the initial regions from different diagonals may be joined together to form an approximate alignment with gaps. Only non-overlapping regions may be joined. The score for the joined regions is the sum of the scores of the initial regions minus a joining penalty for each gap. The score of the highest scoring region, at the end of this step, is saved as the initn score.
After computing the initial scores, FastA determines the best segment of similarity between the query sequence and the search set sequence, using a variation of the Smith-Waterman algorithm. The score for this alignment is the opt score.
One of the few ways to evaluate the significance of such a score is to generate an empirical score distribution from the alignment of many random pairs of sequences having the same lengths as the two compared sequences. From this distribution, the Z-value (the number of standard deviations from the mean) for the alignment score of interest can then be estimated. Importantly, it should not be assumed that the score distribution is normal. Under reasonable assumptions the random score distribution for optimal ungaped local alignments can be proved to follow extreme value distribution (which proved to be significantly different from the normal distribution) [5]. In the current versions of FASTA and BLAST search programs, the evaluation of statistical significance is based upon the extreme value distribution. These evaluations take the form of E-values.
The E-value for a given alignment depends upon it's score as well as the lengths of both the query sequence and the database searched. It is the expectancy of the number of distinct alignments with equivalent or superior score when using a random sequence on this database. Thus, an E-value of five is not statistically significant whereas an E-value of 0.01 is. Scores of near
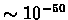 are now seen frequently and they suggest, with extremely high confidence, that the query protein is evolutionary related to the target matched in the database.
are now seen frequently and they suggest, with extremely high confidence, that the query protein is evolutionary related to the target matched in the database.
When the program finds similarity between your query sequence and a database sequence it is not always clear how significant this similarity really is.
To evaluate if this similarity is statistically significance, you can run from the FASTA the package programs prss or prdf [20].
Next: Programs in the FastA3
Up: The FastA Software Package
Previous: Variants of FastA
Itshack Pe`er
1999-01-17