Collecting statistics about amino acids substitution in order to compute the PAM matrices is relatively difficult for
sequences that are distantly diverged, as mentioned in the previous section.
But for sequences that are highly similar, i.e., the PAM divergence distance between them is small,
finding the position correspondence is relatively easy since only few insertions and deletions took place.
Therefore, in the first stage statistics were collected from aligned sequences that were believed to be approximately one PAM
unit diverged and the PAM1 matrix could be computed based on this data, as follows: Let Mij denote
the observed frequency (= estimated probability) of amino acid Ai mutating into amino acid Aj during
one PAM unit of evolutionary change.
M is a
![]() real matrix, with
the values in each matrix column adding up to 1. There is a significant variance between the values in each
column.
For example, see figure 3.1, taken from [4].
real matrix, with
the values in each matrix column adding up to 1. There is a significant variance between the values in each
column.
For example, see figure 3.1, taken from [4].
Once M is known, the matrix Mn gives the probabilities of any amino acid mutating to any other during n PAM units. The (i,j) entry in the PAM n matrix is therefore:
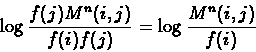
where f(i) and f(j) are the observed frequencies of amino acids Ai and Aj respectively. This approach assumes that the frequencies of the amino acids remain constant over time, and that the mutational processes causing substitutions during an interval of one PAM unit operate in the same manner for longer periods. We take the log value of the probability in order to allow computing the total score of all substitutions using summation rather than multiplication. The PAM matrix is usually organized by dividing the amino acids to groups of relatively similar amino acids and all group members are located in consecutive columns in the matrix.
![\fbox{
\begin{minipage}[h]{\textwidth}
\begin{center}
\begin{displaymath}
\...
...playmath}
\setlength{\unitlength}{0.1000in}
\end{center}
\end{minipage}
}](img9.gif)