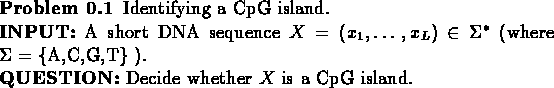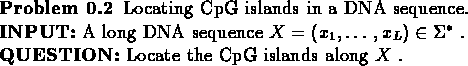CpG is the pair of nucleotides C and G, appearing successively, in this
order, along one DNA strand. It is known that due to biochemical
considerations CpG is relatively rare in most DNA sequences.
However, in particular sub-sequences, which are a few hundred to a few thousand
nucleotides long, the couple CpG is more frequent. These
sub-sequences, called CpG islands, are known to appear in
biologically more significant parts of the genome. The ability
to identify CpG islands along a chromosome will therefore help us spot its
more significant regions of interest, such as the promoters
or 'start' regions of many genes. We will start with the problem of identifying
a given region as a CpG island, and then continue with the problem of locating CpG islands
in a DNA sequence.
We can approach such problems using a Markov chain model.
Let us denote for each
![]() the transition
probability:
the transition
probability:
| (1) |
We assume that ![]() is a random process with a memory of
length 1, i.e., the value of the random variable xi depends
only on its predecessor xi-1. Formally we can write:
is a random process with a memory of
length 1, i.e., the value of the random variable xi depends
only on its predecessor xi-1. Formally we can write:
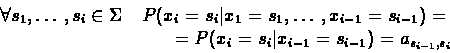 |
(2) |
The probability of the whole sequence X will therefore be:
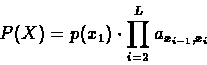 |
(3) |
We can also add fictitious
![]() and
and
![]() symbols to simplify the formula, where
symbols to simplify the formula, where
![]() is the background probability of
the symbol s. Hence:
is the background probability of
the symbol s. Hence:
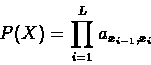 |
(4) |
Let
a+st denote the transition probability of
![]() inside a CpG island and let
a-st denote the
transition probability outside a CpG island (see table
6.1 for the values of these
probabilities, taken from [4]
). We can compute a logarithmic likelihood score for the
sequence X:
inside a CpG island and let
a-st denote the
transition probability outside a CpG island (see table
6.1 for the values of these
probabilities, taken from [4]
). We can compute a logarithmic likelihood score for the
sequence X:
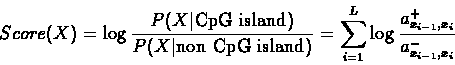 |
(5) |
The higher this score, the more likely it is that X is a CpG
Island.
A naive approach for solving this problem will be to extract a
sliding window
![]() of a given length
of a given length
![]() (where
(where
![]() ,
usually several hundred bases long, and
,
usually several hundred bases long, and
![]() )
to the sequence and calculate
Score(Xk)for each one of the resulting sub-sequences. Sub-sequences that
receive positive scores are potential CpG islands.
)
to the sequence and calculate
Score(Xk)for each one of the resulting sub-sequences. Sub-sequences that
receive positive scores are potential CpG islands.
The main disadvantage in this algorithm is that we have no
information about the lengths of the islands, while the algorithm
suggested above assumes that those islands are at least ![]() nucleotides long. Should we use a value of
nucleotides long. Should we use a value of ![]() which is too
large, the CpG islands would be short sub-strings of our windows,
and the score we give those windows may not be high enough.
On the other hand, windows that are too small might not provide
enough information to determine whether their bases are distributed
like those of an island or not.
A better approach to such problems is described in the following
section.
which is too
large, the CpG islands would be short sub-strings of our windows,
and the score we give those windows may not be high enough.
On the other hand, windows that are too small might not provide
enough information to determine whether their bases are distributed
like those of an island or not.
A better approach to such problems is described in the following
section.