First, practically all the sequences we can obtain today are extracted from extant organisms.
We almost do not know any protein sequences
where one is actually derived from the other. The lack of ancestral protein sequences is handled by assuming that
amino acid mutations are reversible and equally likely in either direction. This assumption, together with the additivity
property of the PAM units derived from its definition, imply that given two amino acid sequences: Si and Sj whose
mutual ancestor is Sij we have:
d(Si,Sj) = d(Si,Sij) + d(Sij,Sj)
when d(i,j) is the PAM distance between amino acid sequences i and j.
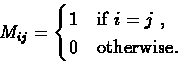
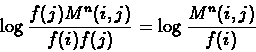
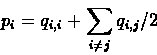
![\fbox{
\begin{minipage}[h]{\textwidth}
\begin{center}
\begin{displaymath}...
...math}
\setlength{\unitlength}{0.1000in}
\end{center}
\end{minipage}
}](img8.gif)
![\fbox{ \begin{minipage}[h]{\textwidth} \begin{center}\input{lec03_figs/lec03_block.eepic} \setlength{\unitlength}{0.1000in} \end{center} \end{minipage} }](img10.gif)
![\fbox{ \begin{minipage}[h]{\textwidth} \begin{center}\input{lec03_figs/lec03_multial.eepic} \setlength{\unitlength}{0.1000in} \end{center} \end{minipage} }](img18.gif)